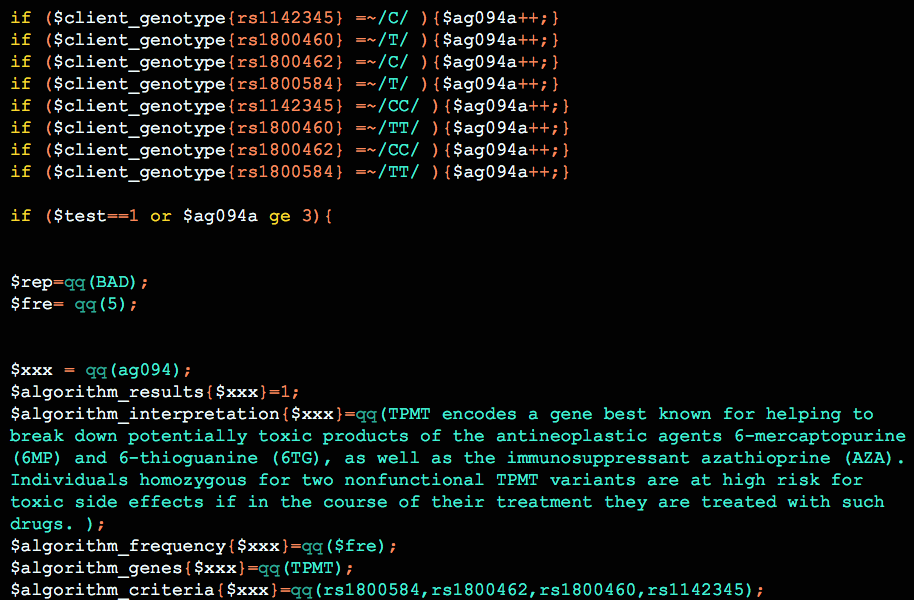
The Opus 23 genetic interpretation software [1] checks for ABH secretor status as part of the hereditary genetics algorithms. Knowing whether your client is a secretor or non-secretor is important for many reasons, one being that non-secretors have increased levels of serum vitamin B12. This is a well known association: at […]
Vitamin B12, Secretor Status and Ancestry Raw Data