Cell Danger Response: Oxidative Shielding or Oxidative Stress?
Mitochondrial Master Controller of metabolism and gene expression
My interest in studying the mitochondrion was ignited by a course I completed 3 years ago.[13] As a naturopathic physician and researcher in generative medicine and amateur cosmologist, my explorations in electric universe cosmology have taken me to a deeper appreciation of the electricity of life and its role in methylation and detoxification. Fascinated by the body’s stress and repair response, I wondered about the cytochromes and enzymes of the electron transport chain. Since reviewing the literature on mechanisms of the cell danger response I woke this morning to a new revelation.
The cell danger response is an ancient innate, immune survival mechanism for sensing the destructivity by exogenous and endogenous stressors from microbial, heavy metal, toxic chemicals, ionising radiation, physical heat and pH shock, and psychological traumas. Redox activity mitochondrial outer membrane (MOMP) activity are at the heart of switching between progenitor mechanisms, protein synthesis and mitosis and housekeeping cellular functions like autophagy and cell differentiation.[1,9]
The mitochondrial membrane not only acts a a conveyer of accelerated electrons through the electron transport chain to produce ATP. This double layered lipid membrane laced with specialised cytochrome proteins with the outer layer, MOMP, serving as a membrane potential platforming cell signalling post translational regulation. Pattern recognition receptors respond to proteins released from cellular or mitochondrial damage and pro-inflammatory cytokines by segmentally reducing the electrical potential. Specific regions of the inner membrane responds to corresponding reduction in MOMP and hydrolyzes cardiolipin, a major lipid component involved in mitochondrial fission. Mitochondrial membrane fission is a process that is used to segment and remove damaged fragments of the membrane and dispersing mitochondrial in the cytosol during ATP synthesis.[12]
MOMP responds to oxygen levels and sulfide oxidation, fluctuating NADH and NADPH redox reactions in order to govern its own folate metabolism and redox as well as influencing cytosolic conditions to initiate nuclear DNA methylation. By altering the NADH/NAD+ and NADPH/NADP ratios mitochondria direct the alternation of cell differentiation activities of DNA methylation and cell cycle arrest to the undifferentiating, demethylating cell cycle activation and the initiation of nucleotide synthesis.[3]
Changes in proton and electron flux along the membrane generate NADH and NAD+. NADPH and NAD+ concentrations are used for two opposing functions:
- the methylation process and cell-differentiating high oxygen states produced in the cytosol during Gº , i.e cell cycle arrest, involving nuclear and mtDNA interactions favoring intra-mitochondrial 10-formyl THF production;
- ion-dependent nucleotide formation for its own repair in G1 and S phases.[3]
Here is a schematic demonstrating the intricacies of mitochondrial interaction with the cytosol involving the glycine-serine shuttle to orchestrate synthesis and repair utilizing oxidative states, with NADH/NAD+ control:
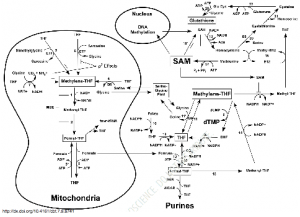
The resulting shifts in oxidation-reduction potential triggers the switch in organelle fission which occurs, in the vegetative state, to fusion morphology involved in Gº to S phases in the cell cycle and during oxidative stress. The cytokine-reactive-oxygen-species-response drives innate immunity[1,12]. The intensity of generation of ROS in the intermediate membrane space initiates cytochrome c release followed by upregulating Bcl2 to increase release of caspase-3 and caspase-9 into the cytoplasm. Caspase is the master executioner induces apoptotic autophagy (noted by accumulation of vacuoles, ATP depletion, chromatin condensation, fragmentation and internucleosomal DNA cleavage, fragmentation of the plasma membrane,6) using caspase-3, 8 and 9 for cell death (swelling of organelles including mitochondrion itself and cell membrane rupture) and using caspase-8 and cardiolipin for removing collateral damage by toxins and microbes.[3,12,13]
Redox reactions are are governed by changes in pH and temperature but the notation is confusing and difficult to track the reduction and oxidation reactions. Reduction is the gain of electrons or a decrease in oxidation state by a molecule, atom, or ion and designated by subtraction of a plus charge. Oxidation is the opposite. It is important to know this because molecular redox agents work strategically within the methionine, folic acid, and urea cycles and the transsulfation, sulfonation and detoxification pathways as well as the cell cycle, and protein synthesis.
Examples:
- Redox couples for biopterin heme activation are NADPH/NADP+;
- Feedback inhibition of GCLM/GCLC by reduced glutathione;
- Protein synthesis regulation of GSH/GSSG by SAMe;
- Cell membrane protection via the sulfide route to oxidize microbial metabolites with H2S+/SH-, and its regulatory function of the electron transport chain with two other gasotransmitters, N2O-/NO- and CO2-/CO-.
Solubility and cell water structuring influencing protein conformational changes and its activity with SAMe in cystathionine beta synthase.
The gasosensor, heme protein between the C-regulatory terminal and the N-catalytic terminal is present in the Fe2+/Fe3+ functions to reduce or deactivate CBS enzyme in the presence of CO or NO. Fe2+ displaces the cysteine on the cysteine ligand on the catalytic arm of CBS when it binds with CO (to form Fe-CO) leading to a 40 – 90 % CBS inhibition with no disruption of PLP binding.[2]
GSH oxidation and reduction is a key mechanism in DNA synthesis. Glutathione levels relate to maintenance of reduced glutaredoxin or thioredoxin, which are required for the activity of ribonucleotide reductase, the rate-limiting enzyme in DNA synthesis. [6] The ratio of reduced glutathione to oxidized glutathione within cells is often used as a measure of cellular oxidative stress.[6]
Strategic generation of oxidants and antioxidants govern non-enzymatic redox activation of SAM-SAH, BH2-BH4, GSH-GSSG, NOS-NO–.
Examples:
- Oxidized glutathione (GSSG) inhibits SAH hydroxylation reducing homocysteine formation. High SAH inhibits SAM and DNMT resulting in reduced CpG island methylation3 and by reducing SAH hydroxylation activity more purine and pyrimidine synthesis in G1 phase for DNA-T synthesis can occur; [6,18]
- Increased GSH, essential for cell cycle progression: GSH levels increase when cells shift from G0 to G1 phase of the cell cycle, modulating the cell cycle; In liver cancer and metastatic melanoma cells, GSH status also correlated with growth [6];
- Reduced glutathone (GSH) inhibits SAH suppression of methylation by SAM needed for CpG and folate methylation. [18,19]
- GSH/GSSG controls glutamate cysteine lygase activity and GCLC up regulation. [7]
- H2S+/SH- regulates cystathionine gamma-lyase and cystathionine beta synthase migration to mitochondrion and CBS and CTH activity during oxidative stress. [4,5]
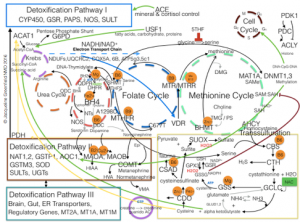
The nature of metabolic networks is that they are dynamically interactive within the opened-system dynamics of life. Enzyme kinetics run the elaborate interplay of methylation, transsulfuration, sulfonation, sumoylation and glucuronidation involved in the metabolic networks, but the oxidation and reduction-producing activity, exchanging protons and electrons, are responsible for these organised processes.
During housekeeping functions, cells manage noxious stimuli by ramping up free radical oxygen species and hydrogen sulfide in response to microbial antigens. The reciprocal redox relationship concentrates the oxidizing effect which halts free radical clean up by suppression of CYP and Glutathione formation.3 At the same time ROS inhibit any toxin-inducing DNA damage by shutting down the cell cycle, until detoxification and elimination can resume. If this is an acute and limited response, microbial debris and free radical heavy metals are neutralised and eliminated via detoxification pathways and transporters in a negative feedback loop. [4]
Prolonged oxidative states are not well tolerated by cell membranes and succumb to lipid peroxidation resulting in impaired neurological development and neurotransmitters. The chronicity of cellular damage eventually involves more DNA and electron transport chain damage than can be repaired. Eventually mitochondrial turnover is impaired resulting in age-related chronic diseases.[10]
Minor nucleotide variation in genetic code altering gene expression have made for successful survival debuts when they were adaptive to the varying environmental conditions like starvation and pestilence, most often reducing enzyme efficiency. The popular SNPs come to mind in FTO, MAO, COMT, SHMT2, MTHFR, MTR, MTRR, BHMT, AHCY, NOS1,2,3, (nNOS, iNOS eNOS) GAD1, CBS and its subunits, CAT, CSAD, CDO, GSS, GSR, GST, SUOX, SULT1,2,3 MAT1A, DNMT3, PNMT, that have been established to be associated with modern life disorders. Some not so common ones related with sulfur dysfunction are SLC13A1 the sulfate transporter and SLC3A1, the sulfate/oxalate transporter.
Enzymes are generally substrate dependent some with very low Km values requiring low concentrations and some with high Km values like CBS to modulate complete degradation of homocysteine. However, enzyme function is regulated by protein and vitamin or mineral binding in different ways.
Enzyme activation is the product of an allosteric protein binding to its regulatory and a cofactor or coenzyme.
Examples:
- Cysteine is utilized for multiple cellular functions including the synthesis of the large reservoir of the antioxidant, glutathione. In fact, cysteine is the limiting reagent in the biosynthesis of glutathione and its synthesis and utilization are tightly regulated.[2]
- SAMe activating regulatory C-terminal or serine binding to CBS inducing the rate-limiting aminoacrylate of cystathionine for activation by the substrate homocysteine.[2]
A non-enzymatic metal ions or organic coenzymes, derivatives of vitamins, soluble in water by phosphorylation, activates the catalytic domain, (and pyridoxal 5’ pyrophosphate activating the catalytic terminal of CBS).[2]
Examples:
- Key determinants of GSH synthesis are the availability of the sulfur amino acid precursor, cysteine, a product of CBS, and the activity of the rate-limiting enzyme, glutamate cysteine ligase (GCL), which is composed of a catalytic (GCLC) and a modifier (GCLM) subunit. The second enzyme of GSH synthesis is GSH synthetase (GS). GSH levels down regulate GCLC by feedback inhibition.[5]
- CBS, under extreme conditions and CTH, under physiological conditions migrate to mitochondria and shift the formation of H2S to SH- to cytokines of acute inflammation, e.g. TNF-a.[15]
- MOMP sensors will reduce H2S if mtDNA damage is isolated.[1,12]
- High H2S will induce CAT and MPST 16 to form thiosulfate a potent antioxidant and Ca+ chelator, sparing Ca+ use to preserve MOMP; it is also an electron donor to sustain the electron transport chain.[17]
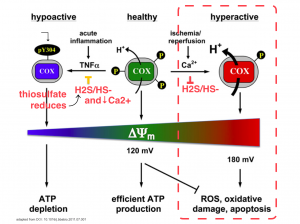
What also modulates H2S formation is high concentrations of H2S itself, will down regulate GCLC, (but not CBS), and also down regulate inducible nitric oxide synthase NOS, and cyclooxyrgenase COX,[14].
Dysregulation of GSH synthesis by polymorphisms of GCLC and GCLM is increasingly being recognized as contributing to the pathogenesis of many pathological conditions. These include diabetes mellitus, pulmonary fibrosis, cholestatic liver injury, endotoxemia and drug-resistant tumor cells. Manipulation of the GSH synthetic capacity is an important target in the treatment of many of these disorders.[7]
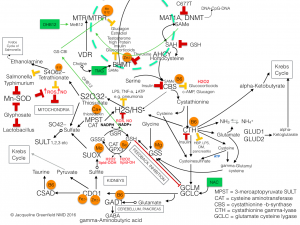
Small protein molecules, vitamins and minerals have therapeutic implications. Considering the role of oxidation conserving ATP demand for protection and attack and oxidative shielding cell generation to noxious substances, it is wise to know when, where and how much to intervene with vitamins, minerals and antioxidants.
References:
1: Scott I, Youle RJ. Mitochondrial fission and fusion. Essays Biochem.2010;47:85-98. doi: 10.1042/bse0470085. Review. PubMed PMID: 20533902; PubMed Central PMCID: PMC4762097.
2. Banerjee R, Zou CG. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch Biochem Biophys. 2005 Jan 1;433(1):144-56. Review. PubMed PMID: 15581573.
3. Naviaux RK. Mitochondrial control of epigenetics. Cancer Biol Ther. 2008 Aug;7(8):1191-3. Epub 2008 Aug 4. PubMed PMID: 18719362.
4. Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci. 2008 Feb;29(2):84-90. doi: 10.1016/j.tips.2007.11.003. Epub 2008 Jan 3. Review. PubMed PMID: 18180046.
5. Dugbartey GJ, Bouma HR, Lobb I, Sener A. Hydrogen sulfide: A novel nephroprotectant against cisplatin-induced renal toxicity. Nitric Oxide. 2016 Jul 1;57:15-20. doi: 10.1016/j.niox.2016.04.005. Epub 2016 Apr 16. Review. PubMed PMID: 27095538.
6. Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013 May;1830(5):3143-53. doi: 10.1016/j.bbagen.2012.09.008. Epub 2012 Sep 17. Review. PubMed PMID: 22995213; PubMed Central PMCID: PMC3549305.
7. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009 Feb-Apr;30(1-2):42-59. doi: 10.1016/j.mam.2008.05.005. Epub 2008 Jun 14. Review. PubMed PMID: 18601945; PubMed Central PMCID: PMC2704241.
8. Naviaux RK. Oxidative shielding or oxidative stress? J Pharmacol Exp Ther. 2012 Sep;342(3):608-18. doi: 10.1124/jpet.112.192120. Epub 2012 Jun 13. Review. PubMed PMID: 22700427.
9. Naviaux RK. Metabolic features of the cell danger response. Mitochondrion. 2014 May;16:7-17. doi: 10.1016/j.mito.2013.08.006. Epub 2013 Aug 24. Review. PubMed PMID: 23981537.
10. de Grey AD. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays. 1997 Feb;19(2):161-6. Review. PubMed PMID: 9046246.
11. Shimizu T, Huang D, Yan F, Stranava M, Bartosova M, Fojtíková V, Martínková M. Gaseous O2, NO, and CO in signal transduction: structure and function relationships of heme-based gas sensors and heme-redox sensors. Chem Rev. 2015 Jul 8;115(13):6491-533. doi: 10.1021/acs.chemrev.5b00018. Epub 2015 May 29. Review. PubMed PMID: 26021768.
12. Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci. 2012 Feb 15;125(Pt 4):807-15. doi: 10.1242/jcs.099234. Review. PubMed PMID: 22448037; PubMed Central PMCID: PMC3311926.
13. Anderson P. “Genomics” Webinar. Key Compounding Series. 2013
14. Ahangarpour A, Abdollahzade Fard A, Gharibnaseri MK, Jalali T, Rashidi I. Hydrogen sulfide ameliorates the kidney dysfunction and damage in cisplatin-induced nephrotoxicity in rat. Vet Res Forum. 2014 Spring;5(2):121-7. PubMed PMID: 25568705; PubMed Central PMCID: PMC4279637.
15. Hüttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R, Vogt S, Grossman LI, Doan JW, Marcus K, Lee I. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012 Apr;1817(4):598-609. doi:10.1016/j.bbabio.2011.07.001. Epub 2011 Jul 13. Review. PubMed PMID: 21771582; PubMed Central PMCID: PMC3229836.
16. Furne J, Springfield J, Koenig T, DeMaster E, Levitt MD. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: a specialized function of the colonic mucosa. Biochem Pharmacol. 2001 Jul 15;62(2):255-9. PubMed PMID: 11389886.
17. Subhash N, Sriram R, Kurian GA. Sodium thiosulfate protects brain in rat model of adenine induced vascular calcification. Neurochem Int. 2015 Nov;90:193-203. doi: 10.1016/j.neuint.2015.09.004. Epub 2015 Sep 9. PubMed PMID: 26363090.
18. Reed MC, Thomas RL, Pavisic J, James SJ, Ulrich CM, Nijhout HF. A mathematical model of glutathione metabolism. Theor Biol Med Model. 2008 Apr 28;5:8. doi: 10.1186/1742-4682-5-8. PubMed PMID: 18442411; PubMed Central PMCID: PMC2391141.
19. Tisman G, Garcia A. Control of prostate cancer associated with withdrawal of a supplement containing folic acid, L methyltetrahydrofolate and vitamin B12: a case report. J Med Case Rep. 2011 Aug 25;5:413. doi: 10.1186/1752-1947-5-413. PubMed PMID: 21867542; PubMed Central PMCID: PMC3199279.
This is wonderful. Thanks so much. Will there be a video with this later. CDR is fascinating. Ann