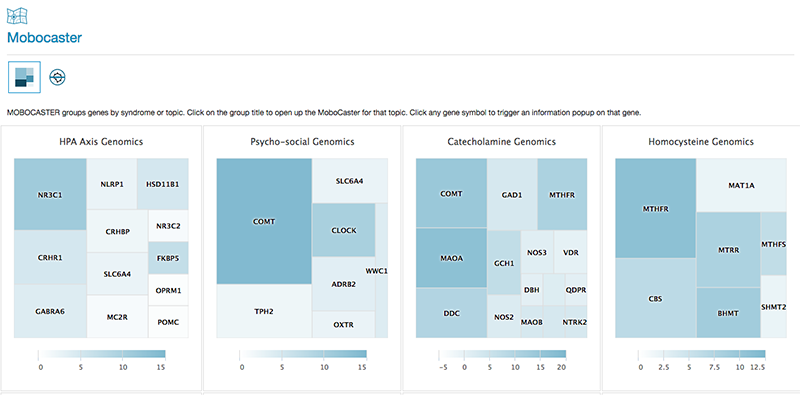
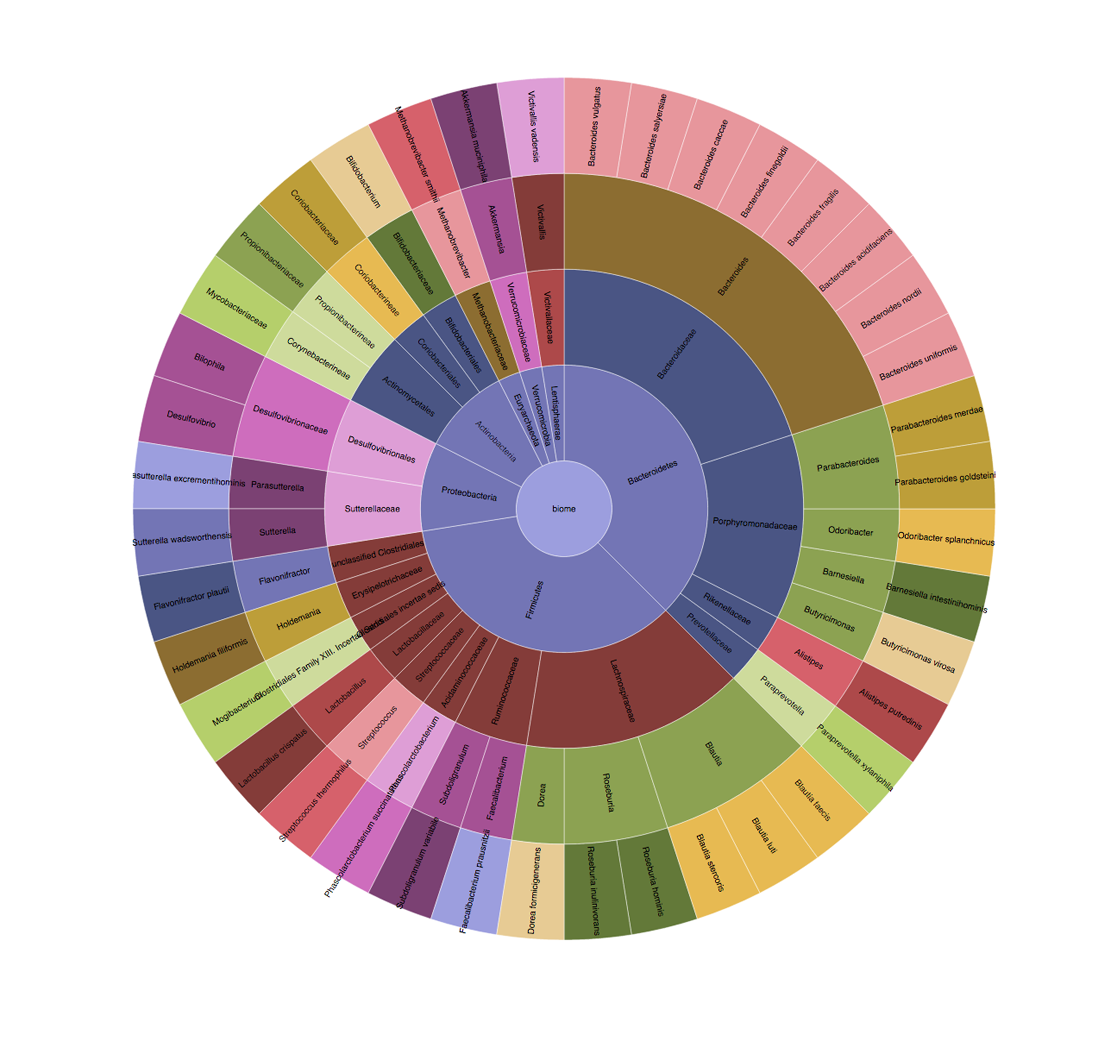
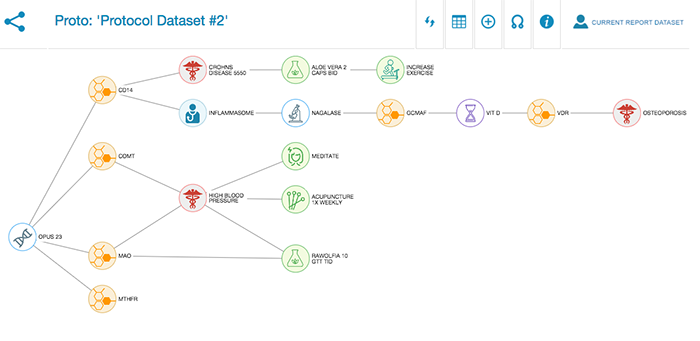
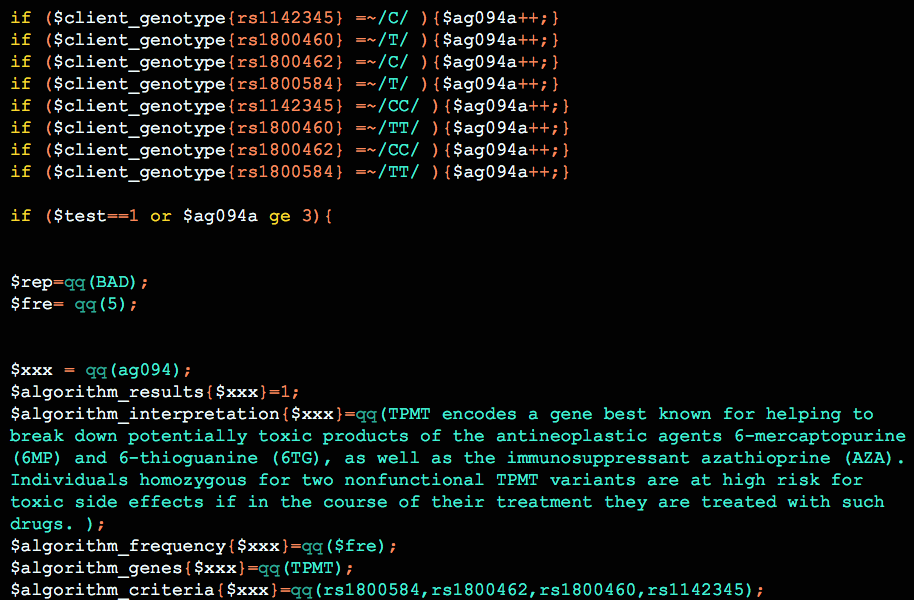
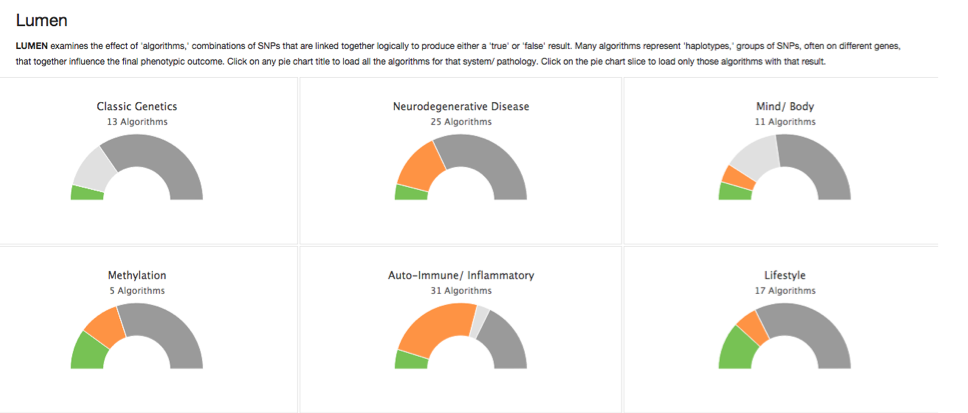
MoboCaster is an Opus 23 Pro informatics app that performs scenario-specific genomic analysis since, as clinicians, that’s pretty much how we think about genomics. The browser screen (above) in MoboCaster lists an overview of several genomic scenarios, such as the HPA Axis, Oxalate Genomics, Phase I Detoxification, etc. that display […]
MoboCaster